News
Contacts:
Steve Bradt
617-496-8070
steve_bradt@harvard.edu
508-856-2000
617-253-1682
617-714-7152
CAMBRIDGE, Mass. – October 8, 2009 - Scientists have deciphered the three-dimensional structure of the human genome, paving the way for new insights into genomic function and expanding our understanding of how cellular DNA folds at scales that dwarf the double helix.
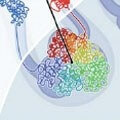
In a paper featured this week on the cover of the journal Science, they describe a new technology called Hi-C and apply it to answer the thorny question of how each of our cells stows some three billion base pairs of DNA while maintaining access to functionally crucial segments. The paper comes from a team led by scientists at Harvard University, the Broad Institute of Harvard and MIT, University of Massachusetts Medical School, and the Massachusetts Institute of Technology.
“We’ve long known that on a small scale, DNA is a double helix,” says co-first author Erez Lieberman-Aiden, a graduate student in the Harvard-MIT Division of Health Science and Technology and a researcher at Harvard’s School of Engineering and Applied Sciences and in the laboratory of Eric Lander at the Broad Institute. “But if the double helix didn’t fold further, the genome in each cell would be two meters long. Scientists have not really understood how the double helix folds to fit into the nucleus of a human cell, which is only about a hundredth of a millimeter in diameter. This new approach enabled us to probe exactly that question.”
The researchers report two striking findings. First, the human genome is organized into two separate compartments, keeping active genes separate and accessible while sequestering unused DNA in a denser storage compartment. Chromosomes snake in and out of the two compartments repeatedly as their DNA alternates between active, gene-rich and inactive, gene-poor stretches.
“Cells cleverly separate the most active genes into their own special neighborhood, to make it easier for proteins and other regulators to reach them,” says Job Dekker, associate professor of biochemistry and molecular pharmacology at UMass Medical School and a senior author of the Science paper.
Second, at a finer scale, the genome adopts an unusual organization known in mathematics as a “fractal.” The specific architecture the scientists found, called a “fractal globule,” enables the cell to pack DNA incredibly tightly – the information density in the nucleus is trillions of times higher than on a computer chip – while avoiding the knots and tangles that might interfere with the cell’s ability to read its own genome. Moreover, the DNA can easily unfold and refold during gene activation, gene repression, and cell replication.
“Nature’s devised a stunningly elegant solution to storing information – a super-dense, knot-free structure,” says senior author Eric Lander, director of the Broad Institute, who is also professor of biology at MIT, and professor of systems biology at Harvard Medical School.
In the past, many scientists had thought that DNA was compressed into a different architecture called an “equilibrium globule,” a configuration that is problematic because it can become densely knotted. The fractal globule architecture, while proposed as a theoretical possibility more than 20 years ago, has never previously been observed.
Key to the current work was the development of the new Hi-C technique, which permits genome-wide analysis of the proximity of individual genes. The scientists first used formaldehyde to link together DNA strands that are nearby in the cell’s nucleus. They then determined the identity of the neighboring segments by shredding the DNA into many tiny pieces, attaching the linked DNA into small loops, and performing massively parallel DNA sequencing.
“By breaking the genome into millions of pieces, we created a spatial map showing how close different parts are to one another,” says co-first author Nynke van Berkum, a postdoctoral researcher at UMass Medical School in Dekker’s laboratory. “We made a fantastic three-dimensional jigsaw puzzle and then, with a computer, solved the puzzle.”
Lieberman-Aiden, van Berkum, Lander, and Dekker’s co-authors are Bryan R. Lajoie of UMMS; Louise Williams, Ido Amit, and Andreas Gnirke of the Broad Institute; Maxim Imakaev and Leonid A. Mirny of MIT; Tobias Ragoczy, Agnes Telling, and Mark Groudine of the Fred Hutchison Cancer Research Center and the University of Washington; Peter J. Sabo, Michael O. Dorschner, Richard Sandstrom, M.A. Bender, and John Stamatoyannopoulos of the University of Washington; and Bradley Bernstein of the Broad Institute and Harvard Medical School.
This work was supported by the Fannie and John Hertz Foundation, the U.S. Department of Defense, the National Science Foundation, the National Space Biomedical Research Institute, the National Human Genome Research Institute, the American Society of Hematology, the National Heart, Lung, and Blood Institute, the National Institute of Diabetes and Digestive and Kidney Diseases, the Keck Foundation, and the National Institutes of Health.
###
Cutting-edge science delivered direct to your inbox.
Join the Harvard SEAS mailing list.