News
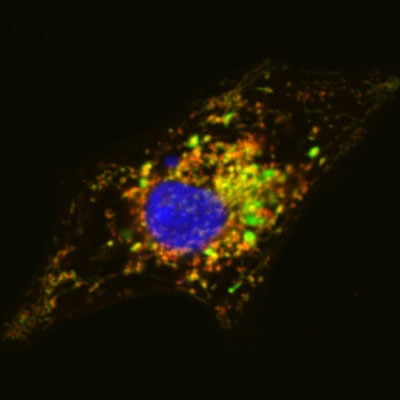
Harvard researchers have corrected a genetic contractile defect in engineered heart tissue in the laboratory. This is the first tissue-based model of correction of a genetic heart disease. (Image courtesy of Gang Wang and William Pu, Children’s Hospital Boston.)
Cambridge, Mass. – May 11, 2014 – Harvard scientists have merged stem cell and “organ-on-a-chip” technologies to grow, for the first time, functioning human heart tissue carrying an inherited cardiovascular disease. The research appears to be a big step forward for personalized medicine, because it is working proof that a chunk of tissue containing a patient’s specific genetic disorder can be replicated in the laboratory.
The work, published in the journal Nature Medicine, is the result of a collaborative effort bringing together scientists from the Harvard Stem Cell Institute (HSCI), the Harvard School of Engineering and Applied Sciences (SEAS), the Wyss Institute for Biologically Inspired Engineering, Children’s Hospital Boston, and Harvard Medical School (HMS). It combines the “organs-on-chips” expertise of Kevin Kit Parker and stem cell and clinical insights by William Pu.
Using their interdisciplinary approach, the investigators modeled the cardiovascular disease Barth syndrome, a rare X-linked cardiac disorder caused by mutation of a single gene called Tafazzin, or TAZ. The disorder, which is currently untreatable, primarily appears in boys, and is associated with a number of symptoms affecting heart and skeletal muscle function.
The researchers took skin cells from two Barth syndrome patients, and manipulated the cells to become stem cells that carried the patients’ TAZ mutations. Instead of using the stem cells to generate single heart cells in a dish, the cells were grown on chips lined with human extracellular matrix proteins that mimicked their natural environment, tricking the cells into joining together as they would if they were forming a diseased human heart. The engineered diseased tissue contracted very weakly, as would the heart muscle of a Barth syndrome patient.
This series of images shows how inserting modified RNA into diseased cells causes the cells to produce functioning versions of the TAZ protein (from left, green), which correctly localize in the mitochondria (red). When the images are merged to demonstrate this localization, green overlaps with red, giving the third image a yellow color around its edges. (Image courtesy of Gang Wang and William Pu, Children’s Hospital Boston.)
The investigators then used genome editing, a technique pioneered by Harvard collaborator George Church, to mutate TAZ in normal cells, confirming that this mutation is sufficient to cause weak contraction in the engineered tissue. On the other hand, delivering the TAZ gene product to diseased tissue in the laboratory corrected the contractile defect, creating the first tissue-based model of correction of a genetic heart disease.
“You don’t really understand the meaning of a single cell’s genetic mutation until you build a huge chunk of organ and see how it functions or doesn’t function,” said Parker, who has spent more than a decade working on organs-on-chips technology. “In the case of the cells grown out of patients with Barth syndrome, we saw much weaker contractions and irregular tissue assembly. Being able to model the disease from a single cell all the way up to heart tissue, I think that’s a big advance.”
Furthermore, the scientists discovered the TAZ mutation works in such a way to disrupt the normal activity of mitochondria, often called the power plants of the cell for their role in making energy. However, the mutation didn’t seem to affect the overall energy supply of the cells. In what could be a newly identified function for mitochondria, the researchers describe a direct link between mitochondrial function and a heart cell’s ability to build itself in a way that allows it to contract.
“The TAZ mutation makes Barth syndrome cells produce an excess amount of reactive oxygen species, or ROS — a normal byproduct of cellular metabolism released by mitochondria — which had not been recognized as an important part of this disease,” said Pu, who cares for patients with the disorder.
“We showed that, at least in the laboratory, if you quench the excessive ROS production then you can restore contractile function,” added Pu, an affiliated faculty member at HSCI and a cardiologist at Children’s Hospital. “Now, whether that can be achieved in an animal model or a patient is a different story, but if that could be done, it would suggest a new therapeutic angle.”
His team is now trying to translate this finding by doing ROS therapy and gene-replacement therapy in animal models of Barth syndrome to see what could potentially help human patients. At the same time, the scientists are using their human “heart disease-on-a-chip” as a testing platform for drugs that are under trial or already approved that might be useful to treat the disorder.
“We tried to thread multiple needles at once, and it certainly paid off,” Parker said. “I feel that the technology that we’ve got ‘arms’ industry and university-based researchers with the tools they need to go after this disease.”
Both Parker, the Tarr Family Professor of Bioengineering and Applied Physics at SEAS, and Pu, who first talked about collaborating while at a 2012 Stockholm conference, credit their partnership and scientific consilience for the success of this research. Parker, who is also a core faculty member of the Wyss Institute and a principal faculty member at HSCI, said that the “organs-on-chips” technology that has been a main effort of his lab only worked as fast and as well as it did because of the high quality of Pu’s patient-derived cardiac cells.
“When we first got those cells down on the chip, Megan [McCain], one of the joint first authors, texted me, ‘This is working,’” he recalled. “We thought we’d have a much harder fight.”
“When I’m asked what’s unique about being at Harvard, I always bring up this story,” Pu said. “The diverse set of people and cutting-edge technology available at Harvard certainly made this study possible.”
Researchers also involved in this work include joint first authors Gang Wang of Children’s Hospital and McCain, who earned her degree at SEAS and is now an assistant professor at the University of Southern California. Amy Roberts of Children’s Hospital and Richard Kelley at the Kennedy Krieger Institute provided patient data and samples, and Frédéric Vaz and his team at the Academic Medical Center in the Netherlands conducted additional analyses. Technical protocols were shared by Kenneth Chien at the Karolinska Institutet.
Topics: Health / Medicine, Bioengineering
Cutting-edge science delivered direct to your inbox.
Join the Harvard SEAS mailing list.
Scientist Profiles

Kit Parker
Tarr Family Professor of Bioengineering and Applied Physics


